Recent work by IBM, featured in articles such as “IBM seeks to simplify robotic chemistry” (by Chemistry World) has made quite a splash recently, with other headlines being “Robotics, AI, and Cloud Computing Combine to Supercharge Chemical and Drug Synthesis” (by IEEE Spectrum) and “IBM has built a new drug-making lab entirely in the cloud” (by the MIT Technology Review).
So what has been done?
When reading through the articles the title chosen by Chemistry World is probably closest to the facts reported by IBM – simplifying synthesis, by using robotics. This, for a compound of interest in drug discovery, which is meant to be made in the lab, involves figuring out a synthetic route (reagents and catalysts to be used), along with optimizing the experimental protocol (solvents, temperature, as well as other reaction conditions). All of this can be a long process, especially for complex molecules, such as where many synthetic steps or stereo centers are involved. And while the extreme end of it, such as complex natural product total synthesis, is sometimes still seen as the ‘pinnacle’ of chemistry (at least historically by some academic groups), in practice this is often a ‘trial and error’ process which one wants to end in a reasonable amount of time, in order to just have the compound available for testing. And this is where robotics, such as the work presented here, can help – by automating synthesis, which can run 24/7, and to allow the chemist to think about the next steps of the project instead. Of course there are a few ifs and buts one should mention when it comes to the practical implementation – and more on that below.
What can we expect, when it comes to drug discovery?
If we now put this into the context of drug discovery, what is our expected impact of using robots for synthesis on the overall process?
To set the scene: An article by Paul et al., “How to improve R&D productivity: the pharmaceutical industry’s grand challenge“, describes two possible scenarios one can be confronted with in a discovery setting – the scenario of few starting points, and the one of many starting points. In pharmaceutical research we generally (but not always!) have an ‘abundance of discovery’, meaning many compounds which hit a target (or are active in a primary assay) and which could potentially taken forward. The question is then how best to proceed with them (and which ones of them!), towards in vivo efficacy and a tolerable safety profile. And this is where disease biology comes into play, be it as a relatively smaller Proof-of-Concept (PoC) study, or a larger clinical trial, as also described by Paul et al. We need to know whether our compound works in vivo – it is comparatively easy to find a ligand (we have millions of them after all in databases such as ChEMBL), but it is comparatively much less easy to find a molecule that is efficacious, and that at the same time possesses a tolerable safety profile, and four out of five compounds entering the clinic currently fail due to one of those two reasons. Given the ‘abundance of discovery’ scenario in drug discovery this is the actual difficulty we encounter – not coming up with new ideas, but rather to select from the abundance of ideas available.
Impact of process improvements on the overall success of drug discovery projects
In relation to the question which improvements to the drug discovery process now would have which type of quantitative impact on project success, we (Isidro Cortes and myself) have recently analyzed the impact of improving speed, quality and cost across the phases of drug discovery (work which is to be published in Drug Discovery Today shortly, it is currently under review in the revised version [update 21 Jan 2021: The articles are currently being published, see http://www.drugdiscovery.net/aireview]).
We asked the questions (which generally follows the analysis in the paper above by Paul et al., with just slightly different analyses and visualizations of results):
If we
- Reduce the time of a discovery phase by 20%; or
- Improve the quality of the decision taken by 20% (i.e., the reduce failure rate in relative terms by 20%); or
- Reduce the cost of the phase by 20%;
Then what is the impact, on average, on the monetary value of a successful drug discovery project at the time of approval (with failures being included in the capital spent, as well as capital costs and the lifetime of patents being considered)?
The result of the analysis is shown here:
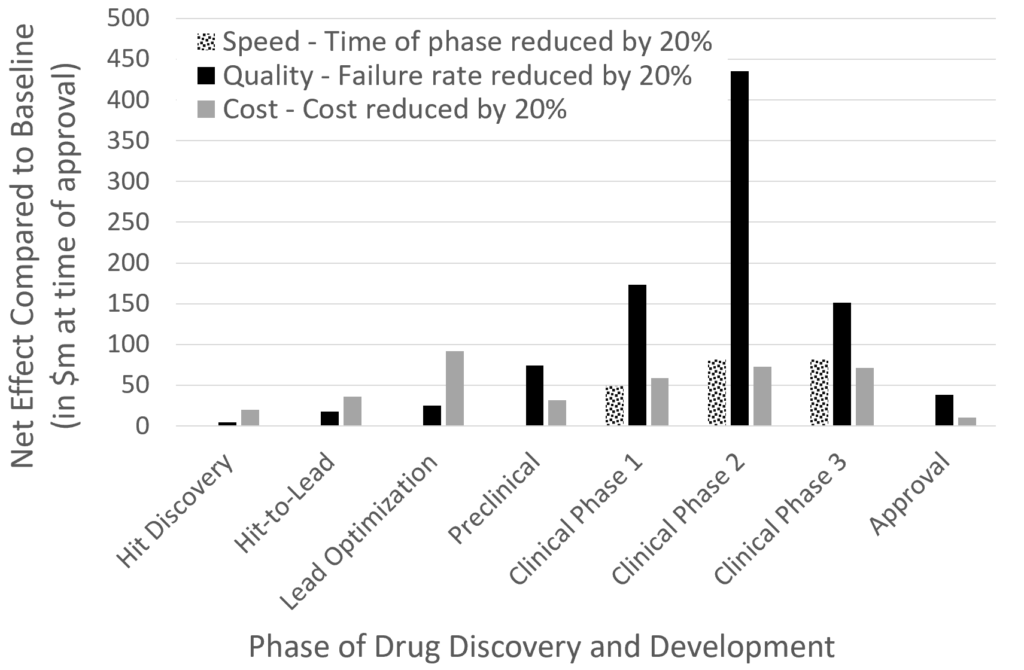
It can be seen that the highest impact can be gained from better decisions (in other words: fewer failures) in the clinical phases by far, followed by reduced time taken in clinical phases, as well as reduced cost in the lead optimization and clinical phases, by approximately similar amounts. (This result is generally in agreement with Paul et al., where we just changed parameters in the simulation in different ways and calculated different output parameters from the model, here summarized as financial impact.) ‘Increasing success in the clinical phase’ has many aspects of course – the compound as much as the disease/disease endotype treated, as well as the combination of the two (and further aspects such as patient recruitment, clinical trial design, etc.)
The bottom line is that increasing success in the clinical phases is of primary importance. However, as can also be seen in the figure, there is also a positive impact of reducing cost during lead optimization, and this is where the current work on automating synthesis (which is heavily involved in lead optimization) would fit in. (This assumes that cost indeed is reduced in the long term; I am not aware of numbers at this stage, and upfront investment is likely significant. However, assuming that some companies such as IBM run ‘synthesis as a service’ this might eventually involve a fee-for-service model, except for big companies who implement such processes fully in-house. Let’s see how things develop.)
Apart from lead optimization another case one where faster synthesis can help though is of course the ‘fail fast and cheap’ paradigm proposed by Paul et al. (which has found relatively widespread adoption since then) – if, but only if we are able to couple faster synthesis with meaningful evidence of biological efficacy and safety. In other words: Making compounds quicker only helps if you know which one to select afterwards. Otherwise you just ‘generate more’ (such as with High-Throughput Screening), but nothing better, which still doesn’t allow you to reach the goal faster, simply since this is not the relevant bottleneck in an ‘abundance of discovery setting’. Becoming faster is very good – as long as we know what to do with this capability. And in this particular case, this means that we need to also have endpoints we can measure which are predictive of efficacy and safety, in order select better in this particular ‘abundance of discovery’ situation. (See also the related paper by Jack Scannell mentioned at the end of this post.)
Unsolved problems
What is frequently neglected in articles written for a general audience are of course the myriad unsolved problems of a scientific approach – which, however, are as important in science as those which are solved already. As every PhD student will experience first hand: One continuously explores borders in science – but, ‘the more you know, the more you know what you don’t know‘, and naturally approaches have their limitations and pitfalls, all of them. (This often, as it certainly did in my case, leads to the subjective impression that one knows less at the end of a PhD than at the start of it, although this is objectively of course very unlikely to be the case.)
In synthesis in particular one should be aware of limitations of data available, in particular of data abstracted from patents (which are naturally designed to confuse the reader and hence difficult to extract); chemical space coverage (or here reaction coverage), and biases in the data (such as the relative lack of data about failed reactions), along with limitations of how models are assessed. (See this article for a recent more detailed discussion of all of those points.) Defining yield is not trivial (when do you say that a reaction ‘worked’? At 1%? At 10%?), substituents might interfere with an otherwise valid mechanism in particular cases, etc. Also technically of course some steps during synthesis and purification are simply difficult to automate, on the purely physical side of things and in the same way combinatorial explosion makes a large number of routes possible, the inability to perform certain steps automatically limits the number of practically available routes drastically.
In the bigger picture human psychology of course plays into the picture in the current direction of science, and our fascination by it- humans like to believe that bigger is better, and large numbers sound impressive (‘we try so often, something simply has to work eventually!’). Equally, at least in ‘Western culture’, there is an underlying believe that ‘technology will solve all of our problems’. But – will it? It obviously depends on the problem at hand and whether it can be solved with technology – and while chemical reactions are predictable with increasing amounts of better data, and improvements in instrumentation, in the area of disease biology we might not be there yet, both due to a lack of understand, and a lack of data. (In this context ‘Can a Biologist Fix a Radio?‘ is a very worthwhile related read.)
This also means that materials discovery is probably quite a different game from drug discovery when it comes to automating discovery – here we stay in the physical domain, which we can understand and model significantly better, and we often have sound theories about underlying determinants of properties. So quite likely advances in automated synthesis and data analysis are, in my personal opinion, more likely to materialize faster than in the drug discovery field, where our understanding of the system, our ability to model it, and to come up with predictive test assays for efficacy and safety endpoints is relatively less developed. Which is, of course, again precisely the selection problem we are confronted with in the ‘abundance of discovery’ setting we are in.
Reflections on the reporting of research in popular media
Both from the titles above, and the articles themselves (such as the one by IEEE Spectrum) it becomes apparent that scientific writers, as opposed to scientists working in the field, sometimes have quite different perspectives on what and how to report about scientific or technological progress. To quote from the above article, in order to reflect on it a bit further: “IBM even had one of the journalists choose the molecule for the demo: a molecule in a potential Covid-19 treatment. And then we watched as the system synthesized and tested the molecule and provided its analysis in a PDF document that we all saw in the other journalist’s computer. It all worked; again, that’s confidence.”
Firstly, I doubt that a journalist suddenly desired to synthesize “1-(3-Bromophenyl)methanamine” – so this needed to be in some kind of pre-defined (and hence limited) list of compounds to synthesize. Secondly, and probably more importantly, this is not really the chemistry present in most drug discovery projects – to look at the compound in all its glory:
This looks more like an industry chemical than a ‘drug’ (though of course such generalizations can be misleading, if one only reviews lists of eg the top-selling drugs, with all the different types of chemistry present). Still, synthetic complexity in this case has almost certainly been lower than what would be needed in practice in most cases when synthesizing a bioactive compound. So the validation of the method hasn’t been chosen with practical relevance in mind.
Thirdly, also from the IEEE article, what is the point of using a ‘potential Covid-19 treatment’ (with a structure with too little complexity to fully make the point) for a case study? This is no treatment of any sort yet (see above comments in in vivo efficacy and safety), so this is a rather misleading claim. Likewise statements such as that the developments reported ‘may have cut the discovery time for Covid-19 treatments in half’ are entirely arbitrary (why not by 51%? or, now that we are at it, 99%?). (Potentially 1-(3-Bromophenyl)methanamine is even the cure for everything of course… but potentially my dog can also fly to the moon. Just, in practice, he doesn’t tend to do it that often.)
The terminology of ‘drug-making in the cloud’ by the MIT Technology Review is equally misleading of course – this setup has, at the current point in time, not produced any drugs. Drug discovery has never been held back by a ‘lack of cloud’ – it has been (and still is!) difficult since in vivo efficacy and safety are hard to anticipate for a new compound… which (as the reader may expect by now) goes back to the selection problem in the ‘abundance of discovery’ setting again.
So in the interest of scientific credibility a bit less ‘cloud computing’ and ‘supercharging drug discovery’ might be desirable- in the end no one is helped by selling unrealistic expectations (except of course tempting readers to click on the article, but I don’t think this justifies making misleading claims either).
Conclusion
The synthesis of small molecules is work-intensive and often involves repetitive work, which makes it very suitable for automation. In particular in the area of optimizing reaction conditions automation may hence have real impact in getting to the goal faster, and this will very likely lead to advances in discovery in particular in physical areas, such as materials discovery.
However, in the drug discovery field the setup is different. Here, in the ‘abundance of discovery’ situation, not coming up with new ideas, but rather selecting the most promising of them – the ones that are likely to be efficacious and safe in vivo – is the most difficult step. This means that we would need a sufficiently close coupling of synthesis with biologically meaningful endpoints that allow us to select compounds better, in order to really make an impact on the field, when it comes to drug discovery which is meant to be clinically relevant.
Finally, in this context a publication that I cannot recommend enough is “When Quality Beats Quantity: Decision Theory, Drug Discovery, and the Reproducibility Crisis” by Jack Scannell (also a YouTube discussion by the author is available). In short: Lower numbers, but better selection criteria, work better to identify a compound with the desired properties than larger numbers, but using less relevant (noisy, or plainly less relevant) selection criteria to pick the winners. The devil is, and continues to be, in the biological domain, and ‘AI’, ‘the cloud’ and technology more generally will only help in a limited way in this situation when it comes to the discovery of novel compounds with desired in vivo properties.
Should we analyze the life science data we have? Absolutely – quite likely we will be able to identify new links in it. Is the research reported in the above article worth it when it comes to synthesis itself? Absolutely – we are able to automate often repetitive processes. However, in order to truly advance drug discovery, when it comes to clinical relevance, we need to see the chemical and the biological aspects of drug discovery in an integrated manner – so we can make compounds more easily, and that those compounds that we decide to make also have a suitable efficacy and safety profile in vivo.
(Many of the above points will also be expanded upon in an upcoming article by Isidro Cortes and myself in Drug Discovery Today; please check the journal website towards the end of September 2020 in case of interest, I will also link it from here when available.)
/Andreas
Pingback: Cambridge Cheminformatics Newsletter, 14 October 2020 – DrugDiscovery.NET – AI in Drug Discovery