Predictive Models – Is There Anything Wrong?
Computational modelling of data (or ‘Artificial Intelligence’ if you prefer) has made huge advances, most recently particularly in areas such as image and speech recognition – and some of those applications have led, or may well lead, to practical applications (just to pick cellular phenotyping as an example). Whether the immense investment also translates into a benefit for society remains to be seen of course.
However, advances in other areas than speech and image recognition, in particular drug discovery, are much more difficult to clearly identify. One particular reason for this is, besides the (small) amounts of (in vivo relevant!) data available, the inconsistency of its labeling (since labels, such as toxicity, or the indication of a drug, etc.) depend on dose, assay setup/disease endotype, etc. (we will get to that in an upcoming Drug Discovery Today Technologies article; I will link it from here when available [update 22 Jan 2021: The articles are now being published in Drug Discovery Today, see http://www.drugdiscovery.net/aireview]).
But another aspect is the way models have been validated, and often are still validated within the domain, and which in my opinion is often somewhere between insufficient, or outright meaningless in the way they are presented in for example scientific publications. But – it is not all up do the individual – model validation, and here we will mainly focus on property models of small molecules, is simply much more difficult than it sounds! Some of the reasons why model validation is difficult are relatively easier to address and only require suitable effort, while some others are domain-specific, and it is very hard to do anything about them (at least without considerable, and often unfeasible, experimental effort that would be required). Still: The more aware we are what the pitfalls of model validations are, the better we can at least do what is possible, which in the medium term will be of benefit for the field, science as a whole, and ultimately society (e.g. by helping drug discovery in a meaningful way, in the real world).
A remark before I am getting into it: I have certainly committed some of the sins outlined below myself – and this is precisely why I feel exceptionally qualified talking about them right now! I will hence use some of my earlier work in this article, and revisit it in the form of a self-critical review, which I believe is also important for myself, to advance my own science. If others can learn from this text as well – then even better.
The Disconnect Between Happy Publications and Sober Reality
When doing my PhD I certainly was part of the ‘we need to pump up those numbers’ brigade – whatever the data set and its biological meaning, from bioactivity (in the virtual screening setting) to toxicity prediction, the aim was to improve recall, accuracy, or whatever other performance measure was desired. And why not? After all ‘pushing the numbers a bit’ ensured a publication, and wasn’t that all that was required? I didn’t pay real attention to the endpoint that had been measured (whether it was only a proxy measure, or had a meaning it its own right for example); the reproducibility of the data; the question to what extent a certain type of data set split translates to prospective performance (!); or the context a model is embedded in (e.g. to make a drug, instead of a ligand, you need to consider properties far beyond on-target activity itself of course), etc. etc.
This problem became even clearer to me when applying models in practice, be it in the academic or applied drug discovery context – the models simply didn’t turn out to be as good as promised by the numbers in previous ‘validations’. So, were those validation then valid, after all? It is now apparent that there is a fundamental underlying disconnect between establishing, and reporting, performance of models on the one hand, and ensuring their suitability for prospective application on the other hand – and both are even existing, as it sometimes appears to me, in parallel universes. This goes beyond pure science of course, for example there are wrong incentives at play (e.g. you need to ‘pump up the numbers’ to be able to publish), but unfortunately this means that the translation into practice (as described in further detail below) nearly necessarily loses out: There is simply a huge conflict between ways scientific ‘progress’ is evaluated when it comes to publications, and what matters in a real-world scenario.
Sounds cryptic? Read on and we’ll go right into it!
So what is the problem when it comes to model generation, and reporting model results, in particular when modelling properties of small molecules?
A handy ‘cheat sheet’ of what we will discuss in the following here as a table, with a convenient dual use: To choose your favorite way of cheating, or as a checklist (be it as an author or a reviewer) to ensure a given piece of work is as good as possible!
| # | Aspect | Why It Matters | Frequent Problems | Addressable? |
|---|---|---|---|---|
| 1 | Data set size, composition, coverage, and training-test split are insufficient | Performance and data used to obtain a given performance are intrinsically related | Data sets are unavailable for comparison; they are biased; insufficiently large/diverse; train/test splits splits chosen are irrelevant for prospective performance | Only very partially (by reporting data for comparison); not fundamentally though, since (a) we cannot describe chemical space, hence cannot sample it or describe any inherent data set biases; and (b) future chemistry to predict for will likely be different from any training/test sets available today |
| 2 | Prospective validation is still generally insufficient to estimate model performance | Predicting new molecules is the proof of the pudding – but only if sufficient in number, distribution and without human intervention! | Too small numbers of molecules tested; manual intervention in selection process; insufficient attention to baseline control (if used at all); extrapolation to different/future chemistry still impossible | In principle, but virtually impossible practically (due to time and expense needed). And: “Model validations are process validations.” |
| 3 | Baseline model not properly chosen, optimized, or compared | A baseline defines advancement made over existing methods | Unclear which baseline method to choose; less incentive to optimize baseline method than new method under development; differences are often not as significant (or relevant) as described | Yes, with suitable effort |
| 4 | Data quality not assessed or taken into account when reporting performance | Models are based on data, and its quality defines the upper limit of model performance | Model performance is reported without paying attention to data quality used for model generation | Data quality cannot be changed (except by generating more/different data), but can (and should be) reported |
| 5 | Irrelevant model endpoints chosen or not discussed in context of actual model application | Model can only be used in practice if the endpoint it models matters for the end goal one aims to achieve | Increase in model performance is reported as an end goal in itself, without discussing relevance of endpoint for decision making in a real-world setting | In principle yes – but frequently large data sets are only available for (easily measurable) proxy endpoints whose impact on decision making is subjective. Also wrong incentives in publication process are a problem (see main text) |
| 6 | Ascribing model impact on complex outcomes is impossible | If a model claims to be used in the drug discovery context then its impact on this endpoint (and not ‘ligand discovery’ etc.) needs to be quantified | It is either possible to evaluate a model in context of early, data-rich, but less relevant endpoints; or in the context of late, data-poor, but relevant endpoints | No. Retrospective evaluations may provide some guidance, but suffer heavily from ‘survivor’ (and other) biases And: “Model validations are process validations.” |
1. Model Performance and Data are Intrinsically Linked – And One Does not Mean Anything Without the Other
While performance of a model is reported as standard, the data set – and its characteristics – usually aren’t
That datasets differ in size and distribution of data points is a given; and (in the chemical field) luckily making datasets available to reproduce results (or obtain results on a given benchmark data set using a new method) has become more common over the ten years or so. However, while the performance of a method is usually characterized with a variety of performance measures, the same is not true for the datasets themselves. This is a problem, since the numerical performance of a method, the data set used to evaluate the method, and its prospective performance are intrinsically related. And if we don’t understand whether the chemical space covered by a given test set is comparable to that of another, or to our chemical space of interest in the future (which we are usually not even aware of ourselves!), then reporting numerical performance of methods is by and large meaningless. (More formally speaking, the data of our sets are not exchangeable.) While this is the case in any area, it is an even bigger problem in the chemical domain, since (a) chemical space is (very large, (b) available data samples it only insufficiently, and (c) future applications will only require predictions for a very small subset of chemical space. In other words: The performance of a given method on a new data set will look (often vastly) different – and you need to start afresh with what you learned regarding the performance of a method. This results in obvious problems.
Why is chemical space so unique? There are many reasons for it – most notably its size, and on the other hand our inability to know anything about the underlying structure of this space: We don’t know whether the chemistry exhibiting bioactivity against a target is similar due to synthesis (or other) bias – or due to the underlying selectivity and activity preference of this particular target. We don’t know which chemistry is more (or less) common among the ca 1063 small molecules that are thought to exist up to a molecular weight of 500. This is different from most other areas, say, biological sequences, where you have the significance of replacing amino acids, such as in case of BLOSUM (or other) matrices – none of this exists in the chemical domain in a meaningful way. We cannot state anything about the underlying data distribution of chemical space – so we don’t know where we are in this space; we cannot describe what a ‘representative sample’ is in any way; and we cannot estimate future performance. (This is the global view on molecular property models – of course this gets put into a different context if one accepts that any models is only a local model, but this is not the way models are usually presented in the current literature.)
Data splits – there is no right way, only differently wrong ways
The question of how to ‘split data’ for validation has been the subjective of intense research, and approaches such as cross-validation, leave-class out and time split validation, etc. exist, with results showing in one study that “time-split selection gives an R2 that is more like that of true prospective prediction than the R2 from random-selection (too optimistic) or from our analog of leave-class-out selection (too pessimistic).” However, does this hold true in any case? I would probably agree that cross-validation overestimates performance, while leave-class out validation underestimates it – if you cross-validate (and assign compound randomly into folds) you sample from the same underlying distribution (leading hence to an overestimate in model performance), while in the leave-class out setting you go to the opposite extreme of predicting far away from your training data set. But: time-split performance often fluctuates, which makes it also difficult to come to a conclusion as to model performance (but which of course may very well lie in the nature of the matter). And why are time splits problematic conceptually? While they do approximate the situation of ‘predicting the future’ during time split you don’t care about the sampling – you assume that going from time t to time t+1 will have an approximately similar effect as going from t+1 to t+2. However, most people would agree with me that going from 2019 to 2020 felt rather different, compared to going from 2018 to 2019 – there is nothing fixed, or constant, about going one time element into the future, when it comes to the data, such as molecules being generated in a pharmaceutical company. A time split is not performed in the chemical domain – so what you assume is that, on average, the novelty/extrapolation power of data splits approaches that in the future. This may be the case in some areas, but probably not in others (e.g. for new modalities). Time-split uses just a random piece of ‘new’ chemical space, of limited size – given we don’t know how representative that is either, we are ‘just’ (in both the positive and negative sense) sampling model performance, from the whole chemical space the model might be applied to in the future. So do I recommend not using time-split validation? Not really – it might very well be the best we can do retrospectively, so among all the wrong ways of validating retrospectively this might well be the least wrong one of all.
(A related quote I quite like regarding time-split validation from a recent paper: “Time-split cross-validation – like all other retrospective methods – can only tell you about how the model will perform if it is never used in the decision-making process.”)
For further related reading the interested reader may be referred to e.g. The Importance of Being Earnest: Validation is the Absolute Essential for Successful Application and Interpretation of QSPR Models, to more fundamental treatment of the subject matter of model selection available.
So should we ‘design’ validation datasets?
One argument to get around ‘biased’ validations that has been made in the past is that we should ‘design’ datasets to remove said biases during model validation (see e.g. MUV , AVE, LIT-PCBA approaches etc.). However, the problem is that we don’t have knowledge of the underlying total chemical space at all, and neither any theoretical basis of the chemical space possessing a certain property, such as activity against targets etc. Hence, should we retain (and during validation ‘re-discover’) close analogues, or not? This is impossible to say; we only have the data available in databases (which huge analogue bias etc.) and cannot estimate in any reliable way how representative the sample is for the underlying distribution. So in my opinion all you do is swap an unknown (implicit) bias with a known (explicit) bias – and this ‘devil you know’ might feel more comfortable than the devil you don’t know. But will this bring us closer to estimating prospective performance now? Evidence rather points to the contrary, that removing ‘biases’ does not improve performance on distant molecules (see e.g. here and here) – and, rather, taking advantage of data biases might be a more practically useful path to take in practice, to bias our models towards identifying molecules with the property of interest. In the end we don’t want a model that is unbiased but doesn’t work – we would probably rather have one that does the job, even at the cost (if it is one in the first place) of bias. From what I can see there is no real way of ‘designing’ ourselves out of the problem of data availability in retrospective validation studies in its entirety.
Core of the problem: Chemical space!
To now come to the core of the problem of the above points: We don’t know what is ‘all chemical space’, what matters, and we cannot sample it (and existing samples are hugely biased). In biology (e.g. for sequence clustering) the situation is quite different – we have mutation probabilities, which we can observe from evolution. In chemistry, we can sample chemical space up to a point (such as up to 17 heavy atoms in GDB-17) – but apart from the molecules being predicted and not covering larger structures we even more importantly do not know what the underlying distribution of data points with a given label of relevance is. So how diverse is the total chemical space binding to a given receptor? We don’t know – some receptors are more promiscuous than others. But we don’t know – so we can only remove bias with respect to an arbitrary viewpoint.
Beware of the numbers
In this section I should include various ‘data enrichment’ strategies which have been reported which lead to increased performance, in particular the addition of negative samples. We have done this before ourselves in the context of targets prediction (where we performed sphere exclusion of inactive compounds to add structures to the negative data set), and there are other studies on e.g. adding stochastic negative examples in the same application area. And, as has been reported in both studies you do improve numerical performance in a cross-validation setting, so isn’t this laudable? Well, not really – since, in the first case, you are making the classification problem simply easier for the classifier if you pick inactives which are by design more dissimilar to actives in the first case. Yes, your numbers get better – but will a prospective application agree? In the latter study the authors report themselves that a temporal split showed markedly less improvement in performance, and in some cases even no improvement at all. Hence, beware: A performance number reported in one context (and retrospective validation) simply cannot be translated to a number reported in another context (of a true, prospective application of the model).
One interesting (but at the same time somewhat trivial) observation is that increasing performance numbers and practical model utility are often anti-correlated – larger performance numbers seem to indicate better models, but at the same time lower coverage of chemical space; while lower numbers explain more diversity in the data, but which results in more difficult datasets, and hence numerically worse models. We pretend to build models with best performance for future applications, but I am not sure we currently validate models in a way to really reach this goal.
Conclusion: Performance reported depends on a particular data set, and numerical performance cannot be compared between datasets. Likewise, there is no real way to estimate prospective performance of models – all our attempts are just about ‘choosing our preferred devil’ when validating models, be it cross-validation, time split validation, etc. We do not understand chemical space, and our sampling for any label is biased in an unknown way, hence we cannot estimate (or remove) any bias present in the data. There is no right way to validate a model – just ‘more’ or ‘somewhat less’ wrong ways.
2. Prospective validation is often still insufficient in practice
One might now be tempted to argue that a prospective validation will tell us how well a model is working – it is predicting the future (unseen compounds) after all, isn’t it! However, there are (at least) four reasons why this sounds easier than it is in practice.:
Firstly, validation sets are often too small – and this is of course due to practical limitations of any study. A better example of our own work (where a somewhat larger validation set was used) is probably Proteochemometrics Modelling for Bromodomains where 1,139 compounds were tested prospectively and where selection followed a standardized protocol to the largest extent possible (though aspects such as compound availability still introduced a significant bias in the validation). A worse example is probably the selection of 50 positive predictions and 10 negative predictions for a hERG model, which is smaller (in particular the inactive set), and where manual selection was involved. Basically, the latter attempt at model validation is more closely related to ‘proof by example’ – and we cannot really say how good the model would be on a large, unbiased sample of chemical space based on this experiment.
The latter validation brings me, secondly, to the point of manual (or real-world) intervention – in many cases selecting the top-ranked examples automatically for validation is not of interest (for example due to a large number of analogues); and even if it is possible, the validation is limited by the compounds available from a vendor or library (or, in the case of virtual library, of the reagents and reactions available etc.) Hence there is always some kind of fiddling with the algorithm predictions involved when moving to experiment. So what is the contribution of the model, and what is the contribution of the manual steps involved after obtaining results back from the experiment? This is simply impossible to say. (One other way to express this is that model validation is always process validation, there is simply no way of validating a model on its own.)
Thirdly, there is the baseline control. In experiments we need a positive and a negative control to put response to a new compound into context. The same is true for a predictive model – and the problems with choosing an appropriate baseline control deserves a separate point below. Here we will only focus on the problems of performing prospective validation for the baseline control: Even if we picked a large number of compounds predicted by the model, in an unbiased fashion, and say spent considerable resources synthesizing (or purchasing) 1,000 compounds predicted to be active for a model, would be do the same for the compounds predicted to be inactive, or a control group? This halves the resources to be spent on the actually interesting part of the project, so this is often (and understandably) not done. But even if we did invest those resources, then our baseline control will necessarily come up with other compounds suggested for testing, which has its own compound availability etc. bias – so even if we wanted to, we were not able to perform a prospective validation that compares a new method and the baseline method in a fair way. But this is not even the worst point, which is probably…
Fourthly, that we still don’t know how representative compounds selected by a method are of future applications of the model. We evaluate on what is known today (be it actives, or known chemistry such as in vendor catalogues, or even virtual libraries, based on known reactions) – but how representative is all that for the next project, a next target class, new chemistry etc.? Due to the size of chemical space, and the impossibility of measuring how well one samples it we have no way of knowing how much we know, compared to what we don’t know. (And no, approaches of plotting what we known in various ways doesn’t cut it, the problem is much more fundamental than that – projecting high-dimensional chemical space into 2D PCA plots is simply unfeasible. Take the dimensionality and the choice of descriptors for a start – and think about the relevance of those descriptors for an arbitrary endpoint next, in a sparse data set. Do you really know which compounds should be plotted next to each other in this representation?) So should we do e.g. a diversity selection for prospective validation instead? This is not really feasible either – which descriptor would you choose, and … do we really care about all chemical space equally? We can gain confidence, we can show for parts of chemical space (the ‘Applicability Domain’, if we manage to define this practically) of a model that it works. After all, maybe this is what we need to become comfortable with – all of our models are in the end just local models, that are numbers assigned on an arbitrarily chosen test or validation set, and this is as much as we can do in practice.
Conclusion: Prospective validation sounds good in theory, but there are real practical and conceptual problems with it. On the practical level, the prospective sample is often either too small, and/or biased, and not representative of any other future applications of the model. This is particularly of relevance given the size of chemical space, and our ability to prospectively validate a meaningful part of this chemical space. On the conceptual level, model validation is always process validation and a model cannot be validated by itself, without the surrounding process of applying the model. Hence, prospective validation is the right thing to do in principle, but very difficult to impossible to perform in practice.
3. Comparison to state of the art is more difficult than it sounds
When publishing a new method we need to compare it to the state-of-the-art in a meaningful way, in order to measure whether true progress has been made. However, this is more difficult than it sounds, be it due to choice of the baseline method in the first place, the care applied to hyperparameter and parameter optimization during model training, as well as the numerical comparison of model performances which may be misleading.
Picking a baseline method is a often by what is generally accepted to be a well-performing method for modelling data – 10 or 15 years ago one might have chosen a Support Vector Machine for this purpose, today probably a Random Forest, and you will have (usually) done a good choice for a method that is frequently used, and that performs well in many situations. Is this the best current models can do though? We only know what we try, so we cannot really say. Also, what is equally useful as a baseline is a very basic method – say, how well does e.g. a mean predictor perform, (partial) linear regression, or ‘dumb features‘ (counts of atoms) in a virtual screening setting? That is a useful negative control to add as well, just to keep us grounded – in this way we will be able to detect whether the complexity of the methods we employ is actually justified, by increased performance compared to the very simple baseline. So we can conclude that there is generally a degree of arbitrariness involved in selecting a baseline method, and that we need to ensure if the complexity of any of the methods employed are justified by performance improvements over any very simple baseline. Only if employing a new method is justified in both regards we can truly talk of progress when reporting a supposedly superior performance of any new new method or model.
However, things get worse when training the baseline method – since, of course, hyperparameter exploration and parameter optimization is needed here, and with the same rigor as for the new method under development. However, is this really done for optimizing a ‘bog standard’ Random Forest model, when the novel method is so much more exciting, and one needs this improved performance for a publication? Here, in the bigger picture, incentives for publications (and academic career progress etc.) might simply get into the way of the best science possible. Still – the baseline must be allowed to be as good as possible, to really claim an advance of a new method or model, compared to the existing state-of-the-art, otherwise there will not be a fair comparison.
And once the baseline method has been obtained, how does one compare it to the new method under development? Of course science is quantitative, so ‘proof by example’ won’t do – but do we really pay attention to practical relevance of improvements in model performance, or only statistical significance (and if the latter, do we really calculate this in the right way)? It should be stated explicitly here that statistical significance does not imply practical relevance – in case of large numbers of samples rather small effect sizes can become significant, so both aspects of model performance need to be considered individually.
Let us turn to a concrete example of current interest: Does, say, deep learning outperform other methods in bioactivity prediction, or not? Deep learning is sometimes performing better in this area than other methods (but not always); and it has been pointed out that attention needs to be paid to how performance is evaluated. So what does a proper performance evaluation comprise from the numerical side (even beyond other aspects of model validation discussed elsewhere in this article)? At the very least paying attention to the right performance measure (in this case, use of Precision-Recall Curves is advocated in addition to ROC curves; for further characteristics and limitations of ROC curves see this work), and ensuring significance of changes in performance (if this is what one is after), given the size and number of datasets available. In this particular study it has been found that, in a target prediction setting, Support Vector Machines give rather comparative performance to deep learning methods under the criteria for performance comparison employed.
As an aside on deep learning methods, in other areas again the (small) amounts of data available may render such methods nonviable – and the general limitations of ‘pushing the numbers’ on proxy endpoints are discussed in point 6 of this article. My personal feeling (from both direct experience and available literature) is that, yes, there is quite likely a value in deep learning for large datasets, and that also transfer learning seems to improve performance in particular for small datasets. Still, how much better are we really – and does this really matter in the drug discovery context, and to what extent? I am overall skeptical to what extent predicting proxy measures better numerically will really improve our ability to make decisions in the drug discovery context.
Finally, one needs to be careful how results are presented – e.g. in a recent study on analyzing Electronic Health Records with deep learning the abstract describes the virtues and performance of this particular method used, but omits that the confidence interval of the performance of the logistic regression baseline overlaps with the deep learning method (see last part of SI for the baseline information) – so even if the information of a baseline method is available, one needs take care not to put any subjective spin on the numbers one obtains (… which of course brings us back to the many wrong incentives driving scientific publications again).
Conclusion: We are only able to determine whether we make progress beyond the state-of-the-art if we compare a new method or model thoroughly to what exists already. This involves in particular the choice of baseline method, its parameter and hyperparameter optimization, as well as a suitable quantitative approach of comparing model performance. Only if all of those aspects are fulfilled we are able to state that a new model is better than a previous one (and that is within the limitations described elsewhere in this article).
4. Not considering data, data quality, and the context of a model
Generating a model, validating it (to the extent possible), and publishing it/making it available is of course not an end goal in itself – we don’t just want an algorithm to ‘give us numbers’, we want predictions which we can use to make decisions, and in many cases those predictions are mean to be used either in place of, or at least to prioritize, subsequent experiments. And as any experimentalist will be aware: (a) Data comes with an error; and (b) assays come with assay conditions. What does this mean for us? It means that (a) there is an upper limit of model performance that we can expect from a model (the model cannot be better than the data used to generate it); and (b) there is not only one way to ‘generate a model’ for an endpoint, but there are rather many ways to experimentally determine endpoints, and often they do not agree with each other. As an example from the bioactivity space take kinase inhibition, which can be measured in various different types of assays and with different ATP concentrations etc. (see this review for details). But also something as apparently ‘trivial’ as solubility depends heavily on the setup chosen, where kinetic and thermodynamic solubilities, and hence ways of measuring this endpoint exist. Even more so, complex biological endpoints such as DILI come with different annotations which makes their modelling non-trivial. We will leave the second point – of choosing practically meaningful assay endpoints – to item 5. in this list, and focus here on the variability/error of an assay which is inherent to the particular assay endpoint chosen.
Of course, error in the data can be quantified – say in cytotoxicity data or public databases such as ChEBML and so on. One can then put maximum model performance into the context of assay reproducibility, which has been commented on already more than a decade ago, and also for regression models in detail more recently. Just: This is hardly ever done when model performance is reported in the literature, be it due to neglect, or since this is often difficult to do in practice (e.g. due to lack of replicates). This, however is a major shortcoming of any predictive method which does not consider the error in the data. Experiments give you a value, and an associated uncertainty of a measurement. In the same way, a model also needs to give you a value, and an associated uncertainty of this prediction – and the latter cannot be detached from the uncertainty of the experimental measurement.
Conclusion: Both measurements and computational predictive models provide a value, and an associated uncertainty of the measurement (or prediction). Given that a predictive model is based on experimental data, it also needs to take into account the uncertainty of the underlying measurement it uses for model generation.
5. Relevant and irrelevant models – “It’s more important to do the right thing, than to do things right”
In some sense this is probably the most important part of the generation of computational models; what is the endpoint we decide to model in the first place, and is it actually of relevance for the real-world questions asked?
And here a core problem comes up, that is frequently not even mentioned in scientific publications: Endpoints are treated as ‘the real gospel’, as if they meant something by themselves in every case. However, this is far from true – often experiments are designed for endpoints not because they are of the utmost relevance in drug discovery, but because they are somehow associated with relevant endpoints, and they are easily measurable. Take for example logD, which is associated with solubility, permeability, metabolism (and hence essential PK properties of a molecule). and which is hence an important parameter which determines behaviour of a small molecule. But is this property used for decision-making just by itself? Of course not – the whole context of the particular project (e.g. desired PK to reach a particular target tissue etc.) as well as other properties of the compound need to be taken into account for any decision that will be made. So, say, a computational model for logD (or any other proxy endpoint) is just that: “a model of a model” (to quote Graham Smith of AstraZeneca) here – i.e. a computational model for a proxy endpoint.
This has profound implications in practice: Many of the currently published studies of using ‘AI/ML in drug design’ are validated by optimizing numbers for proxy endpoints – but how does this then translate to any practical decision-making? Is an improved prediction of a proxy endpoint by a few percent with say a huge neural network really helpful in practice? The impact of the current (third; after the 1980s, and 2000) hype of computational approaches to drug discovery is still not really apparent, in particular in relation to the resources invested – and a focus on modelling proxy data (and, in turn, shying away from complex biological question) is in my personal opinion quite a likely contributor to that. Turned on its head: Quite possibly we would do better by understanding the question better, and generating the right type of data in the first instance- instead of getting stuck with proxy endpoints forever, and choosing such endpoints to claim victory when it comes to AI in drug discovery in many instances.
Ligand discovery is simply not drug discovery – and from predicting on-target activity and logD it is just a long way to efficacy and safety in vivo.
Choosing ‘wrong’ endpoints is not really a lie, I should add here – but it is at the least overclaiming the relevance of the model, by removing it from its context. As particular examples, again from my own work, I would like to point to models we generated for drug-likeness, for drug-disease associations and compound synergy, which I would like to critically assess here myself. Why do those endpoints modeled need to be understood in context? Well, drug-likeness is based on purely historic evidence of which drugs have been discovered (and where often a large amount of serendipity was involved, even if neat diagrams of ‘drug discovery processes’ try to tell you that this is a completely rational process; the discovery of penicillin may be a nice history example). So the drug space we know is not based on any underlying fundamental laws of nature; it is a chemical space that is biased by chance, and which in addition ‘moves’ considerably, both due to new chemistry aiming at difficult-to-drug proteins, and completely new modalities, such as oligonucletodies (ASOs), PROTACs, etc. However, modelling ‘drug-likeness’ means simply describing what has been done historically, thereby – if applied in the wrong way – hampering future discoveries outside the box. (I should add that all what Chris Lipinski claimed was that the majority of compounds with particular structural properties are available orally, with explicit reference to exceptions for active transport etc. However, the concept was used, and also abused, in various ways subsequently – and modelling ‘drug-likeness’ as an endpoint is likely less useful than modelling say oral bioavailability, which has a physical basis. See also ‘Inflation of correlation in the pursuit of drug-likeness‘ for a more detailed discussion of the topic.) As for drug-disease associations, similar caveats apply: Datasets are necessarily those which are known already, we don’t know ‘false false positives’ in particular, which are of actual key interest of a model, meaning the new drug-disease associations which haven’t been known before. Hence, performance numbers generated by any such model will necessarily emphasize (and be optimized towards) ‘re-discovery’, and not discovery of something new (which will in fact decrease numerical model performance!). So here the problems are less in the concept (although factors such as dose etc. are insufficiently considered as well), and more in the data available to really validate such a model, even on the conceptual basis. Finally, what is not so trivial about modelling synergy? The problems with synergy are that firstly (and as summarized in recent articles) the definition depends on the null hypothesis one (by and large arbitrarily) defines, meaning that synergy mathematically derived from a dose-response matrix of two compounds using one null hypothesis can even be interpreted as antagonism in another (!). Furthermore, if we consider practical utility of such a model, data can reasonably only be obtained on cell lines; so how does any synergy finding translate to the in vivo situation (where disease is more more heterogeneous, compounds are available at different concentrations, etc.)? This is a particularly difficult point since synergy is usually dose-dependent, and hence PK of compounds in a combination really matters in practice. Due to all of those reasons this makes the modelling of cell-line derived synergy values using one arbitrary synergy annotation simply less relevant in practice. It’s not only about the numbers, also in the cases discussed here – and the embedding of the model will define whether the model will have a meaning in the end, or not.
What of course often happens is that fields evolve to adopt early benchmark data sets – and subsequently those benchmark datasets are accepted as ‘state of the art’ and required for benchmarking. ‘Competitions’ have a similar effect on the field. The problem with this is that any limitations of the accepted benchmark data set become less important, and the field moves on to ‘increase numbers’ as their primary objective – which, however, does not necessarily imply increased practical utility of a model.
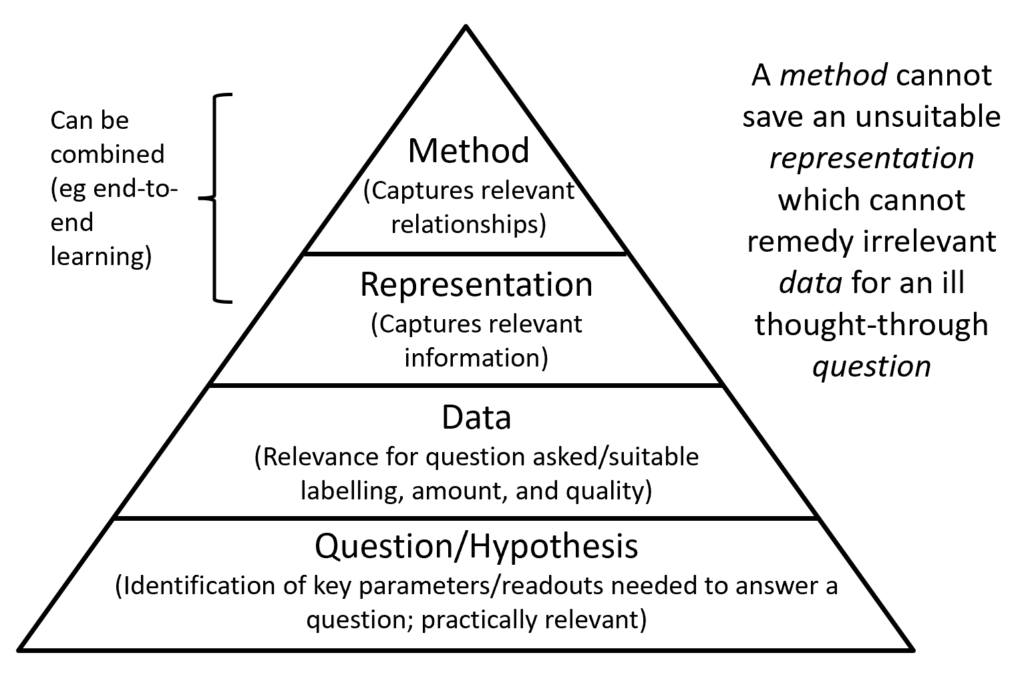
Conclusion: “It is more important to do the right thing, than to do things right” – which means, we firstly need to find the right endpoints to model, which needs to be followed by the identification of suitable data, an appropriate representation, and only finally the appropriate modelling method. This is visualized in the figure above – and only in this way we will reach the final goal of AI having a meaningful impact on drug discovery. Improving the prediction of proxy endpoints numerically is on the other hand less likely to achieve this.
6. Defining contribution of a computational method to project success
And finally, let’s imagine we have worked on all of the above items, with an enormous budget and sufficient time available. A compound that was, say, selected, or designed de novo in the computer, has become a drug (or validated in other ways in an in vivo setting)! Can we say what the contribution of the algorithm was, in this particular case? This is virtually impossible to do – the list of choices has simply been very long to reach this stage (see preceding items of this article for some of them). In which way did compound availability influence the compounds taken forward? Other human interventions? Has really been a suitable baseline control been performed? All of those points would need to be addressed though to be sure about the contribution of a computational model to overall project outcome. A recent paper phrased it succinctly in this way: “It is critical to emphasize that prospective studies validate processes and not models.”
Summary
To come back to the above list, and to summarize either ‘how to lie best’, or of course also to to describe which pitfalls to watch out for in one’s own work (or when reviewing papers):
| # | Aspect | How to lie best | Will I be found out? | What can be addressed? |
|---|---|---|---|---|
| 1 | Data distribution and split | Select data set/split which gives best performance | Medium- would need to be reproduced by others | Sufficient characterization of data set; using different splits |
| 2 | Prospective validation | Pick examples you know which work (selective reporting would be the other option) | Impossible (the referee/reader will not be able to judge hypothetical other model outcomes) | Choosing prospective examples on a large scale and in as unbiased a way as possible |
| 3 | Use of baseline model | Pick baseline which looks plausible but performs badly (and don't optimize model parameters properly) | Only if data set provided; otherwise the referee/reader will not be able to judge hypothetical other model outcomes | Choice of suitable baseline models, paying as much attention to their performance as to the main model |
| 4 | Data quality | Ignore any aspects related to data quality | Only if explicitly asked for - not apparent from model performance itself | Assessing uncertainty in data and maximum possible model performance. Taking uncertainty into account in modelling process |
| 5 | Modelling irrelevant endpoints | Ignore the point, and just 'pump up the numbers' | If the referees and readers are aware of the way the model is used in a real-world situation then yes, otherwise no | Considering both the model itself, and its embedding in a real-world process embedding |
| 6 | Ascribing outcome of process to model | Don't mention alternatives and leave contribution of model as the only plausible choice why a process succeeded | Unlikely - readers or referees are unable to re-implement alternative model scenarios and to investigate other | Addressing the points above properly, and discussing other possible contributions to process outcome |
So… where are we?
It might have become apparent that there is no easy solution here – a true gold standard of ‘validating a model’ is not easy (and, in the absolute sense, even impossible) to achieve. Still, some aspects probably can be addressed by practitioners in the field, in order to make models derived as relevant to real-world applications in the drug discovery context as possible. This means for example that data sets can and should be provided where possible; they need to be characterized to put any derived model performance into context of the data set; different and suitable data set splits should be performed to estimate model performance in at least an approximate way; prospective validation should be as large and unbiased as possible; a baseline model should be used and sufficient attention be paid to its parameter and hyperparameter optimization; and data quality and real-world model use should be discussed along the model performance of the particular endpoint being modeled. Finally, when ascribing the outcome of a process to a model care and critical self-review needs to be in place.
We should use data, absolutely – it would be foolish not to make use of what we already learned in the past. But if we get stuck with the current all we do is the ‘my number is higher than yours game’, then all of us lose out in the end, in the longer term (since resources of society are not used in the best possible way). Credibility is important for the experimental/modeller interface, see what happened to QSAR/CADD in and after its first peak in the 1980s. We need to be realistic what is possible and what is not, be it related to data, methods or other aspects of what we do.
Of course there are also underlying driving forces that push into the opposite direction, such as the ‘need’ for academics to publish in ‘high impact factor journals’, which leads to a ‘prestigious grant’, and vice versa (repeat ad infinitum), Bigger driving forces in society, such as the current hype of AI, and using metrics in all walks of life, equally don’t really help – but all of those points will deserve their separate blog posts in the near future.
I would like to conclude by repeating two quotes I very much like. and which have been used several times above, from a recent publication:
“Time-split cross-validation – like all other retrospective methods – can only tell you about how the model will perform if it is never used in the decision-making process.”; and
“It is critical to emphasize that prospective studies validate processes and not models.”
/Andreas
Acknowledgements: I would like to thank Ant Nicholls (OpenEye) for the initial discussion subsequently leading to this article, at the RSC-BMCS / RSC-CICAG Artificial Intelligence in Chemistry in Cambridge in 2019. All opinions expressed here (and possible errors or oversimplifications etc.) are my own though.
Edit: 14/10/2020 – Spelling of Ant Nicholls’ name has been corrected
Pingback: Cambridge Cheminformatics Newsletter, 14 October 2020 – DrugDiscovery.NET – AI in Drug Discovery
Several other publications/sources of relevance to the topic I have come across more recently after the above article was published:
On Over-fitting in Model Selection and Subsequent Selection Bias in Performance Evaluation
https://www.jmlr.org/papers/volume11/cawley10a/cawley10a.pdf
Recommendations for evaluation of computational methods
https://www.ncbi.nlm.nih.gov/pmc/articles/PMC2311385/
Curing Pharma: (1) Avoiding Hype-based Science
https://www.eyesopen.com/ants-rants/curing-pharma-avoiding-hype-based-science
And another one:
Metrics for Benchmarking and Uncertainty Quantification: Quality, Applicability, and a Path to Best Practices for Machine Learning in Chemistry
https://arxiv.org/abs/2010.00110