Morgan Thomas is a 2nd year PhD student at the Centre of Molecular Informatics at the University of Cambridge in the group of Andreas Bender, funded by and in collaboration with Sosei Heptares. Morgan completed his MChem in Pharmaceutical Chemistry before conducting three rotations in Structural & Analytical Chemistry, Computational Chemistry and Bioinformatics as part of the AstraZeneca Graduate Programme. He is now interested in AI methods for GPCR-based drug design, where his work comparing scoring functions for de novo molecule generation via deep generative molecules was recently published. Morgan is currently working with Sosei Heptares on further extension of the approaches, and applications to GPCR structure-based drug design (SBDD). We interviewed the author to explain the motivation and key findings behind his work.
Congratulations on your recent publication! Could you please summarize what you have done in this work, and in which way this is relevant for drug discovery?
Morgan Thomas (MT): Thank you very much! In this work, the underlying aim was simple, to compare the use of either a structure-based scoring function or a ligand-based scoring function to guide de novo molecule generation by a deep generative model. We felt that there was a real need for this type of research, because current approaches tend to only use one or the other (with fewer published examples of structure-based scoring functions), without truly assessing the practical applicability for drug discovery projects. In fact, when we started this work there was no evidence yet that structure-based scoring functions could be optimized. The majority of current research is focussed on new generative model approaches, or new optimization algorithms, instead of the effect of the scoring functions employed – which however, as we show, have a large impact on de novo molecule chemistry that is being generated. Furthermore, our collaboration with Chris de Graaf, Head of Computational Chemistry at Sosei Heptares, and his team put us in an ideal place to explore the application of structure-based scoring function with these generative AI approaches in particular in the context of GPCR drug discovery.
We addressed this question by using the REINVENT algorithm – a SMILES-based language model – to optimize the predicted activity against Dopamine Receptor D2 (DRD2), for which many annotated ligands for retrospective evaluation are available in databases, and which represents a common current benchmark system. This algorithm uses reinforcement learning for generative model optimization. We optimized two models, one to minimize the docking score determined using Glide and the DRD2 crystal structure (the ‘Glide-Agent’) and one to maximize the predicted probability of activity determined by a support vector machine (SVM) trained on known DRD2 active and inactive ligands (the ‘SVM-Agent’). What we then compared was the behaviour of the generative models according to a suite of benchmarking metrics, a more detailed comparison of de novo molecule chemistry than in other works, and we in particular also looked into predicted protein-ligand interactions with the DRD2 receptor, which in purely ligand-based studies is generally not being done.
And what did you find? Did the ligand-based and the docking-based agent show different behaviour according to those measures?
MT: Firstly, we found that the Glide-Agent was able to minimize the docking score beyond known DRD2 ligands, as shown in the figure below – evidencing that docking score is not too complex a scoring function to optimize. In doing so, the generative model also behaved better than the SVM-Agent with respect to established benchmarking metrics. However, these are only numbers, so we also looked at the chemistry itself that was generated – and here we also found better coverage of known active ligands by the Glide-Agent than the SVM-Agent, likely due to an inherently larger intrinsic diversity of chemical space generated. This was particularly encouraging since, as opposed to ligand-based agents, this type of information wasn’t provided to the model in the first place. There was also many novel chemotypes generated with high docking scores, though. This means that target structure alone provides enough information to find a diverse array of active chemotypes by a generative model, which has in particular implications in situations where no ligands are known at all at the start of a project. From the structural side, the Glide-Agent learned to generate molecules that satisfy residue interactions known to be crucial for ligand affinity against DRD2 – providing a measure of confidence that the Glide-Agent learned relevant information that had previously been experimentally validated.
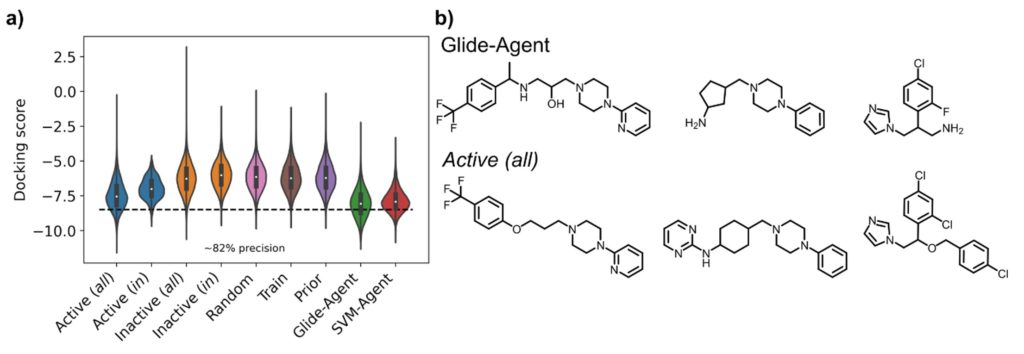
So how does this go beyond what has been known before, how does it advance the field?
MT: On the one hand there is clearly the contribution to including more practically relevant scoring functions in de novo generative models, such as here to find novel chemistry active on a target, and to move away from optimizing less relevant proxy scores. Also we emphasize the importance of the actual chemistry, and ligand-protein interactions, more than other studies, which is only possible due to our explicit use of structure-based information.
By comparing the structure- and ligand-based approaches directly, we show the complementarity in the chemistry obtained using the same generative model, with the same underlying training data. We believe this is predominantly due to the ability of docking to assess also novel chemistry that is not contained in scoring functions used by ligand-based agents, and where predictive models become increasingly less accurate the more distant the predicted chemotype is from the training data. Some methods have trained a predictive model on top of docking (i.e., to predict the docking score of molecules) to speed up evaluation times. However, in our opinion this negates the main advantage of assessing novel areas of chemical space that docking has. Instead, our future work will focus on more efficient optimization algorithms so that fewer evaluations are necessary. This will ensure evaluation is not biased by what has been found before already, which is important for scientific reasons (such as scaffold hopping), as well as from the IP perspective.
So were there any additional findings originating from this work?
MT: Aside from the key questions addressed in this work, we also found that current key benchmarking measures for de novo methods have shortcomings with respect to training data and analysis of chemical diversity. We hence proposed a modification to a key benchmarking data set used to train generative models and proposed a new diversity metric for use in this context. This metric, which we termed ‘Sphere Exclusion Diversity’ (SEDiv), showed better correlation with chemical intuition than currently used metrics, which are for example heavily confounded by molecular weight. We hope these are considered by the community in future work.
Another aspect often overlooked is applying good practice to docking protocols. Many generative models do not account for stereoisomer and protonation state information by default; this may be implicit in QSAR models, but one must account for these explicitly during docking. In this work, we enumerated several possible states of each molecule, and used the best docking score achieved to annotate the structure. The importance of this step translates to other proposed benchmarks which embed a molecule in 3D space irrespective of stereoisomer or protonation state, which however can be crucial for certain binding motifs, e.g. in the context of the current study the ionic interaction with Asp1143×32 in DRD2. Properly preparing ligands for docking can improve enrichment significantly, hence paying attention to this step is good practice in virtual screening and therefore, it should also be considered appropriately in generative model scoring functions.
What was the most difficult part of this work, and what did you do to address the problem?
MT: The most difficult part I found was the evaluation of resulting chemistry and effort towards validating an approach without actually synthesising compounds and measuring binding affinity experimentally. Every approach has its caveats, and every result has a different interpretation, depending on context. For example, validating diversity metrics is very difficult because there is no ‘ground truth’ with respect to molecular diversity, therefore, we had to calculate values for different subsets and align that with chemical intuition, which is completely intangible and still subjective. Likewise, project context defines whether we care more that molecules are very similar to a small proportion of known actives, or broadly similar to much larger proportion. Many such evaluations are therefore subjective to the context of the potential prospective application, and one cannot properly validate a method without this context being known.
Another difficulty of this work was the wall clock time required for docking simulations. As mentioned previously each ligand underwent preparation via enumeration of potential stereoisomers, tautomers and charge/protonation states (which we capped to 8). This meant that a batch size of 64 molecules could result in 64 ligand preparation processes and up to 512 variants for docking simulations. To tackle this computational bottleneck, we parallelised the docking over numerous CPUs. Despite this, training the model for 3000 steps on ~40 CPUs took just shy of one week due to the docking simulations required. Therefore, we are now looking into more efficient optimization algorithms than implemented in REINVENT and faster structure-based scoring functions.
How can the results you have obtained be used by others?
MT: All the data used and generated in this work is available alongside our preprint (for journal reference see below), while the code is freely available on GitHub (https://github.com/MorganCThomas/MolScore). We also uploaded an open-source alternative to Glide (as Glide requires a license) using the Gypsum-DL ligand preparation framework and Smina implementation of Autodock Vina. In fact, the code has much greater functionality than covered in our preprint and can be used with most goal-directed generative models. It is designed to integrate scoring functions together flexibly into an automated framework. We encourage people to try it out, get in contact and contribute other scoring functions / generative models implementations.
Thank you for this conversation, and all the best for your further research!
Further reading:
Morgan Thomas, Rob Smith, Noel M. O’Boyle, Chris de Graaf, Andreas Bender. Comparison of Structure- and Ligand-Based Scoring Functions for Deep Generative Models: A GPCR Case Study. J. Cheminf. 2021, 13 (39) https://doi.org/10.1186/s13321-021-00516-0.
/Andreas
Pingback: Cambridge Cheminformatics Newsletter, 20 May 2021 – DrugDiscovery.NET – AI in Drug Discovery